原标题:NEJM官方翻译|我国团队洛匹那韦-利托那韦医治重症COVID-19
北京时刻 3 月 19 日,《新英格兰医学杂志》(NEJM)在线宣布我国团队关于运用洛匹那韦/利托那韦医治重症新冠病毒病(COVID-19)的临床实验成果。这是新冠病毒疫情爆发以来,世界尖端医学杂志初次宣布医治 COVID-19 的临床实验成果,也是在包含 SARS 在内的近 20 年新发感染病疫情期间宣布的寥寥无几的药物临床实验成果。同期配发的 NEJM 社论称誉该临床实验为勇敢之举。
论文首要作者来自中日友爱医院、国家呼吸病临床研讨中心以及武汉金银潭医院。 全文翻译来自《NEJM医学前沿》。
摘要
布景
现在尚无任何经证明可有用医治 SARS-CoV-2 所造成的重症疾病的办法。
办法
咱们在确诊 SARS-CoV-2 感染(患呼吸体系疾病 COVID-19)而且呼吸周围空气时氧饱和度(SaO2)≤ 94% 或许氧分压(PaO2)与吸入氧浓度(FiO2)的比值 < 300 mmHg 的成人住院患者中展开了一项随机、对照、敞开标签的实验。咱们以 1 : 1 的份额将患者随机分组,别离承受惯例医治联合 14 日的每日两次洛匹那韦-利托那韦(别离为 400 mg 和 100 mg)医治或许独自惯例医治。首要结尾是至临床情况改进的时刻,其界说为从随机分组至 7 分等级量表评分改进 2 分或许出院(以先发作的一项为准)的时刻。
成果
合计 199 例实验室确诊 SARS-CoV-2 感染的患者被随机分组;99 例被分配至洛匹那韦-利托那韦组,100 例被分配至惯例医治组。在至临床情况改进的时刻方面,洛匹那韦-利托那韦医治与惯例医治无差异(临床改进的危险比,1.24;95% 置信区间[CI],0.90~1.72)。洛匹那韦-利托那韦组和惯例医治组的 28 日逝世率类似(19.2% vs. 25.0%;差异,-5.8 个百分点;95% CI,-17.3~5.7)。两组在各时刻点可检出病毒 RNA 的患者百分比类似。在改进意向医治剖析中,与惯例医治比较,洛匹那韦-利托那韦医治使至临床情况改进的中位时刻提早了 1 天(危险比,1.39;95% CI,1.00~1.91)。洛匹那韦-利托那韦组的胃肠道不良事情发作率较高,但惯例医治组的严峻不良事情发作率较高。13 例患者(13.8%)因不良事情提早中止洛匹那韦-利托那韦医治。
定论
在重症 COVID-19 成人住院患者中,与惯例医治比较,咱们未调查到洛匹那韦-利托那韦医治有利。未来对重症患者展开的实验或许有助于承认或扫除该计划发作好处的或许性。(由我国国家严峻新药创制科技严峻专项等赞助;我国临床实验注册号为 ChiCTR2000029308。)
洛匹那韦-利托那韦医治重症 COVID-19 成人住院患者的实验
A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19
DOI: 10.1056/NEJMoa2001282
从 2019 年 12 月开端,一种名为 SARS-CoV-2 的新式冠状病毒在全球引发了称为 COVID-19 的呼吸体系疾病的爆发。COVID-19 的完好疾病谱规模是从轻度自限性呼吸道疾病到重度进行性肺炎、多器官衰竭和逝世。迄今为止尚无针对冠状病毒感染的特异性医治药物。
2003 年严峻急性呼吸体系综合征(SARS)呈现后,对已获赞同的药物进行挑选后发现,洛匹那韦(人类免疫缺点病毒 1 型的天冬氨酸蛋白酶按捺剂)对引起人类 SARS 的 SARS-CoV 病毒具有体外按捺活性。
利托那韦与洛匹那韦联用可经过按捺细胞色素 P450 来延伸洛匹那韦的血浆半衰期。2004 年宣布的一项敞开标签研讨提示,与仅承受利巴韦林医治的前史对照组比较,利巴韦林加用洛匹那韦-利托那韦(别离为 400 mg 和 100 mg)下降了 SARS 患者的不良临床结局(急性呼吸困顿综合征或逝世)危险和病毒载量。
但是,由于上述研讨未进行随机化,无同期对照组,而且联用了糖皮质激素和利巴韦林,因而难以评价洛匹那韦-利托那韦的作用。类似地,在体外实验和动物模型中,洛匹那韦对中东呼吸体系综合征冠状病毒(MERS-CoV)均有活性,而且有病例陈述指出洛匹那韦-利托那韦与利巴韦林和干扰素(IFN)α 联用可铲除病毒,使患者存活。但是,现在尚无的确数据证明将该疗法用于人体的作用,因而医治 MERS 的一项临床实验(联用重组干扰素 β-1b)现在正在进行中(在 ClinicalTrials.gov 注册号为 NCT02845843)。
为了评价口服洛匹那韦-利托那韦医治 SARS-CoV-2 感染的作用和安全性,咱们在 COVID-19 成人住院患者中展开了一项随机、对照、敞开标签实验 LOTUS China(在我国展开的洛匹那韦按捺 SARS-Cov-2 实验 LopinavirTrial for suppression of SARS-Cov-2 in China)。
办法
患者
咱们依据当地疾控中心(CDC)或指定确诊实验室检测出的呼吸道样本 SARS-CoV-2 逆转录-聚合酶链反应(RT-PCR)(上海之江生物科技股份有限公司或圣湘生物科技股份有限公司)阳性成果评价患者是不是满意参加本实验的规范。
本实验的归入规范如下:年纪 ≥ 18 岁,确诊标本的 RT-PCR 成果呈阳性,胸部印象学查看确诊肺炎,呼吸周围空气时氧饱和度(SaO2)≤ 94% 或氧分压(PaO2)与吸入氧浓度(FiO2)的比值(PaO2 : FiO2)≤ 300 mmHg 的男性和非妊娠期女人患者。
扫除规范包含医生以为参加实验不契合患者的最佳利益,患者有导致其无法安全遵从实验计划的情况,已知对洛匹那韦-利托那韦过敏或超敏,已知患重度肝病(例如肝硬化,丙氨酸转氨酶水平 > 5 倍正惯例模上限或天冬氨酸转氨酶水平 > 5 倍正惯例模上限),正在服用与洛匹那韦-利托那韦有配伍忌讳而且在实验期间不能替换或停用的药物,处于妊娠期或哺乳期或许已知有 HIV 感染(原因是忧虑在不联用其他抗逆转录病毒药的情况下,患者对洛匹那韦-利托那韦发作耐药性)。无法吞咽的患者经过鼻胃管承受了洛匹那韦-利托那韦给药。
实验规划和监管
这是一项于 2020 年 1 月 18 日至 2020 年 2 月 3 日(最终一例患者的入组日期)在我国湖北省武汉市金银潭医院展开的敞开标签、个别患者随机对照实验。由于实验紧迫,咱们未能制备洛匹那韦-利托那韦的安慰剂。咱们以 1 : 1 的份额将契合参加实验规范的患者随机分红两组,别离承受为期 14 日的惯例医治联合每日两次洛匹那韦-利托那韦(400 mg 和 100 mg,口服;由国家卫生组织免费供给)医治或独自承受惯例医治。
依据患者的需求,惯例医治包含吸氧、无创和有创通气、抗生素、血管加压药、肾脏替代疗法和体外膜氧合(ECMO)。作为呼吸衰竭严峻程度的目标,为了平衡两组之间氧气支撑的散布情况,咱们依据患者入组时的呼吸支撑办法对随机分组进行了分层:无氧气支撑或许选用鼻导管或面罩的氧气支撑,或许高流量氧无创通气,或许包含 ECMO 在内的有创通气。置换区组(每个区组 4 例患者)随机序列(包含分层)是由一名未参加本实验的统计学家使用 9.4 版 SAS 软件(SAS Institute)预备。为了尽量减小分配偏倚,咱们选用交互式网络应对体系对分组情况做掩蔽,直至经过核算机或电话在该体系上完结随机分组。
本实验取得了金银潭医院组织审查委员会的赞同。咱们从一切患者,或许患者病况过重的情况下从其法定代表取得了书面知情赞同。本实验依照《赫尔辛基宣言》和世界协调会议《药物临床实验质量管理规范》的准则施行。作者担任规划实验以及汇总和剖析数据。作者确保数据的完好性和准确性,并确保实验对实验计划的依从性。
临床和实验室监测
在第 0 日至第 28 日、患者出院或患者逝世期间,由训练有素的护理每日两次依据日记卡评价患者情况,日记卡中记载了 7 分等级量表数据和安全性数据。安全性由金银潭医院的药物临床实验质量管理规范办公室督查。其他临床数据选用 WHO-ISARIC(世界卫生组织-世界严峻急性呼吸体系和新发感染联合会病例记载表进行记载)。
咱们在第 1 日(服用洛匹那韦-利托那韦之前)及第 5、10、14、21 和 28 日(直至患者出院或逝世)收集了患者的口咽拭子样本,并在上海观合医药科技有限公司(泰格和迪安的合资公司)进行了实时定量 RT-PCR 检测(拜见弥补附录)。咱们使用 MagNA Pure 96 体系从临床样本中提取 RNA,并使用 LightMix Modular Wuhan CoV 检测法(TIB MOBIOL),经过 Cobas z480 qPCR(罗氏)检测和定量 RNA。
咱们在每个时刻点均从 199 例患者中的一切仍存活患者收集了这些样本。并未因某一时刻点的拭子检测成果呈阴性而中止之后的样本收集。对基线咽拭子进行了 E 基因、RdRp 基因和 N 基因检测,对之后的样本进行了 E 基因定量和定性检测。临床数据记载在纸质病例记载表中,之后由实验人员两次重复输入电子数据库并进行承认。
结局目标
首要结尾是至临床情况改进的时刻,其界说为从随机分组至 7 分等级量表改进 2 分(与随机分组时的情况比较)或至出院的时刻,以先发作的一项为准。临床情况改进结尾已在咱们之前的流感研讨中运用过,也是 WHO 研制蓝图(R&D Blueprint)专家组引荐的结尾。等级量表在之前已被用作重症流感住院患者临床实验的结尾。7 分等级量表包含以下等级:1. 未住院,且可持续从事日常活动;2. 未住院,但无法持续从事日常活动;3. 住院医治,不需求吸氧;4. 住院医治,需求吸氧;5. 住院医治,需求经鼻高流量氧疗、无创机械通气或这两者;6. 住院医治,需求 ECMO、有创机械通气或这两者均需求;7. 逝世。
其他临床结局包含在第 7 日和第 14 日时选用 7 分等级量表评价的临床情况、28 日逝世率、机械通气持续时刻、生计者的住院时长以及从医治开端至逝世的时刻(天数)。病毒学目标包含随时刻推移,检出病毒 RNA 的患者份额以及病毒 RNA 滴度曲线下面积(AUC)测定值。
安全性结局包含医治期间发作的不良事情、严峻不良事情和提早中止医治。不良事情依据美国国立癌症研讨所《不良事情通用术语规范》(National Cancer Institute Common Terminology Criteria forAdverse Events)4.0 版进行分级。
统计学剖析
发动本实验是对 COVID-19 突发公共卫生事情做出的敏捷应对,其时关于 COVID-19 住院患者临床结局的信息十分有限。开始设定的总样本量为 160 例患者,原因是假定惯例医治组至临床情况改进的中位时刻为 20 天,而且有 75% 的患者将到达临床情况改进,则 160 例患者的样本量将使本实验以 80% 的统计学成效在 α = 0.05 的双侧显着性水平检测出两组的中位时刻有 8 天的差异。依照原计划进行的 160 例患者的入组作业很快完结,但其时断定本实验的统计学成效缺乏;因而决议由研讨者持续归入患者。之后,在另一种药物(瑞德西韦,remdesivir)可用于临床实验之后,咱们决议暂停本实验的患者入组作业。
首要作用剖析是在意向医治的基础上进行,包含承受了随机分组的一切患者。咱们在一切患者到达第 28 日之后评价了至临床情况改进的时刻,并将第 28 日之前未到达临床情况改进或逝世的患者在第 28 日右删失(假如一个人在承受调查的最终时刻之后或许发作了事情,但发作事情的具体时刻不知道,则进行右删失)。
经过 Kaplan–Meier 曲线制作至临床情况改进的时刻,并经过时序查验办法来进行比较。经过 Cox 份额危险模型核算危险比和 95% 置信区间。有 5 例被分配到洛匹那韦-利托那韦组的患者并未承受洛匹那韦-利托那韦医治(其间 3 人在 24 小时内逝世),但被归入了意向医治剖析,由于惯例医治组中并无相应的患者被扫除的情况。咱们还进行了一项扫除 3 例前期逝世患者的改进意向医治剖析。过后剖析包含对英国国家前期预警评分 2(National Early Warning Score 2,NEWS2)≤ 5 分或 > 5 分进行的亚组剖析,以及对发病后 ≤ 12 天或 > 12 天承受随机分组的患者进行的亚组剖析。
由于统计学剖析计划未规则对非必须结局或其他结局查验进行多重性校对,因而咱们将成果陈述为点估量值和 95% 置信区间。置信区间宽度未进行多重性校对,因而不能将置信区间用于揣度关于非必须结局的清晰作用。安全性剖析是依据患者实践承受的医治。咱们选用 9.4 版 SAS 软件进行了统计学剖析(SAS Institute)。
成果
患者
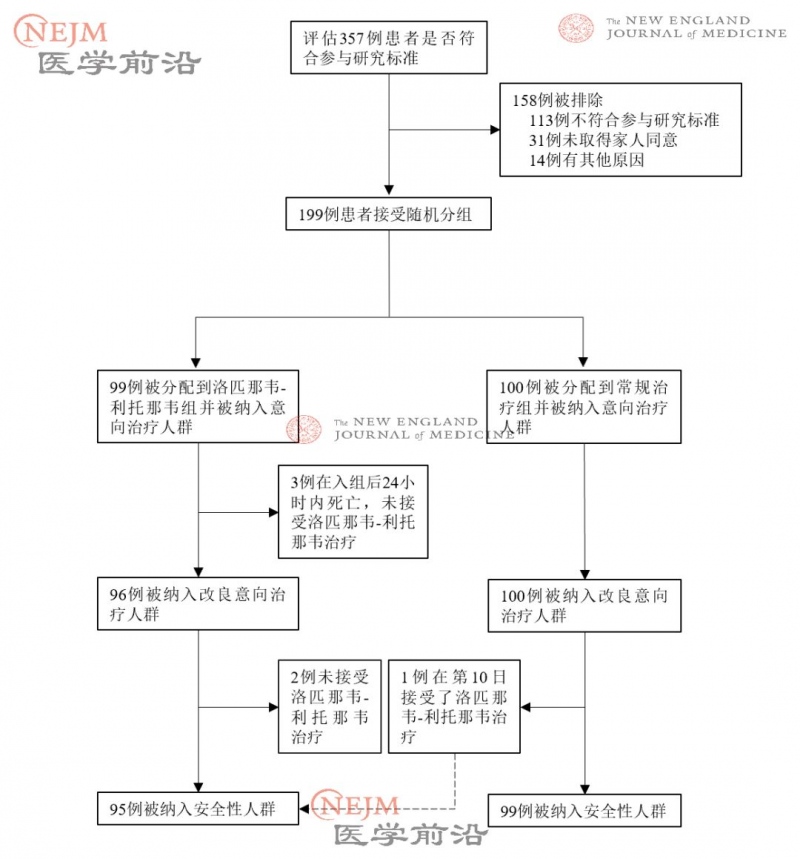
在被随机分组的 199 例患者中,99 例患者被分配承受洛匹那韦-利托那韦医治,100 例患者被分配承受独自惯例医治。在被分配到洛匹那韦-利托那韦组的 99 例患者中,94 例(94.9%)承受了分配的医治(图 1)。在洛匹那韦-利托那韦组中,5 例患者未承受洛匹那韦-利托那韦医治,其间 3 例的原因是在随机分组后 24 小时内前期逝世,别的 2 例的原因是随机分组后,主治医生回绝开出洛匹那韦-利托那韦。
图1. 随机化和医治 胸片分配。
患者中位年纪为 58 岁(四分位距IQR,49~68 岁),60.3% 的患者为男性(表 1 )。呈现症状和随机分组的中位间隔时刻为 13 天(IQR,11 ~16 天)(表 2 )。在人口统计学特征、基线实验室查看成果、等级量表评分散布情况或入组时的 NEWS 2 评分方面,两组间无重要差异。在本实验期间,洛匹那韦-利托那韦组 33.0% 的患者和惯例医治组 35.7% 的患者承受了全身糖皮质激素医治。
表1. 患者基线人口统计学和临床特征。*
* 表中列出的数值是依据取得的数据。咱们取得了洛匹那韦-利托那韦组 95 例患者的白细胞计数、淋巴细胞计数、血小板计数、乳酸脱氢酶和肌酸激酶的实验室查看值;取得了该组 96 例患者的血清肌酐、天冬氨酸转氨酶和丙氨酸转氨酶的数值。咱们取得了惯例医治组 99 例患者的血清肌酐、天冬氨酸转氨酶、丙氨酸转氨酶、乳酸脱氢酶和肌酸激酶的实验室查看值。将肌酐值转换为 mg / dL 需除以 88.4。IQR 标明四分位距。
表2. 患者归入实验时或归入实验后的情况和承受的医治。*
* 加减值为均值 ± SD。ECMO 标明体外膜氧合,HFNC 标明经鼻高流量氧疗,NEWS2 标明英国国家前期预警评分 2。
首要结局
在意向医治人群中,洛匹那韦-利托那韦组和惯例医治组患者至临床情况改进的时刻无差异(中位数,16 天 vs. 16 天;临床改进的危险比,1.31;95% 置信区间[CI],0.95~1.85;P = 0.09)(图 2 )。
在改进意向医治人群中,洛匹那韦-利托那韦组和惯例医治组患者至临床情况改进的中位时刻别离为 15 天和 16 天(危险比,1.39;95% CI,1.00~1.91)(弥补附录表 S1 和图 S1 )。
在意向医治人群中,症状呈现后12日内承受洛匹那韦-利托那韦医治与较早到达临床情况改进相关(危险比,1.25;95% CI,1.77~2.05),而较晚承受洛匹那韦-利托那韦医治则与较早到达临床情况改进不相关(危险比,1.30;95% CI,0.84~1.99)(图 S2A 和 S2B)。
在意向医治人群中,假如依据入组时的NEWS2 评分对至临床情况改进的时刻进行评价,未调查到显着差异(图 S3A 和 S3B)。此外,两组之间至临床情况恶化(界说为 7 分量表评分加剧 1 分)的时刻无差异(临床改进的危险比,1.01;95% CI,0.76~1.34)(图 S4)。
图2. 意向医治人群中至临床情况改进的时刻。
非必须结局
在意向医治人群(19.2% vs. 25.0%;差异,-5.8 个百分点;95% CI,-17.3~5.7)或改进意向医治人群(16.7% vs. 25.0%;差异,-8.3 个百分点;95% CI,-19.6~3)中,洛匹那韦-利托那韦组的 28 日逝世率在数值上均低于惯例医治组(表 3 )。
洛匹那韦-利托那韦组患者的重症监护病房(ICU)住院时刻比惯例医治组患者短(中位数,6 天 vs. 11 天;差异,-5 天;95% CI,-9~0),而且从随机分组至出院的时刻在数值上比惯例医治组短(中位数,12 天 vs. 14 天;差异,1 天;95% CI,0~3)。此外,第 14 日时,洛匹那韦-利托那韦组中临床情况改进的患者百分比高于惯例医治组(45.5% vs. 30.0%;差异,15.5 个百分点;95% CI,2.2~28.8)(图 S5 )。其他结局(例如吸氧持续时刻、住院时长和从随机分组至逝世的时刻)无显着差异。
表3.意向医治人群的结局。*
* 临床情况改进的界说为在修订版 7 分等级量表上,临床情况下降 2 分或许出院。ICU 标明重症监护病房。
† 差异标明为率差或许中位差(Hodges–Lehmann估量)和 95% 置信区间。
‡ 使用 Cox 份额危险模型预算临床改进的危险比。
§ 这一总数包含在随机分组后 24 小时内逝世,因而未承受洛匹那韦-利托那韦医治的 3 例患者。
病毒学
在确诊性呼吸道样本的 RT-PCR 检测成果呈阳性的患者中,合计 69 例(35%)患者的咽拭子样本(获取知情赞同后收集的样本)RT-PCR 检测成果呈阴性。随机分组时,洛匹那韦-利托那韦组患者咽拭子样本(获取知情赞同后收集的样本)的基线病毒 RNA 载量均匀值(± SD)略高于惯例医治组(4.4 ± 2.0 log10 仿制 / mL vs. 3.7 ± 2.1 log10 仿制 / mL)(表 2 )。洛匹那韦-利托那韦组患者和惯例医治组患者随时刻推移的病毒RNA载量无差异(图 3 ),包含依据患病时刻进行的剖析(图 S6 )。
图3. 经过qPCR测出的咽拭子标本SARS-CoV-2病毒RNA载量相关于基线的均匀改变。
I 条标明 95% 置信区间。小于聚合酶链反应(PCR)检测定量下限而且大于定性检测限的成果用 1 log10 仿制 / ml 添补;病毒 RNA 阴性的患者成果用 0 log10 仿制 / mL 添补。在 199 例患者中,130 例(洛匹那韦-利托那韦组 59 例和惯例医治组 71 例)有用于核算病毒载量的病毒学数据,而其他患者在此期间的咽拭子未检出病毒 RNA 。
在任何采样日,洛匹那韦-利托那韦组和惯例医治组可检出 SARS-CoV-2 病毒 RNA 的患者百分比均类似(第 5 日,34.5% vs. 32.9%;第 10 日,50.0% vs. 48.6%;第 14 日,55.2% vs. 57.1%;第 21 日,58.6% vs. 58.6%;第 28 日,60.3% vs. 58.6%)(表 S2 )。
安全性
洛匹那韦-利托那韦组 46 例患者(48.4%)和惯例医治组 49 例患者(49.5%)在随机分组至第 28 日期间陈述了不良事情(表 4 )。洛匹那韦-利托那韦组中胃肠道不良事情(包含厌恶、吐逆和腹泻)的发作率高于惯例医治组(表 4 )。两组中有实验室查看成果反常的患者百分比类似(表 4 )。51 例患者发作了严峻不良事情:洛匹那韦-利托那韦组 19 起和惯例医治组 32 起(表 4 )。洛匹那韦-利托那韦组发作了 4 起胃肠道严峻不良事情,而惯例医治组未发作胃肠道严峻不良事情;研讨者断定这 4 起事情均与实验药物相关。惯例医治组患者的呼吸衰竭、急性肾损害和继发感染发作率高于洛匹那韦-利托那韦组。研讨中心的研讨者断定调查期间的一切逝世均与干涉无关。
表4. 安全性人群中的不良事情总结。*
* 表中列出了随机分组后至第 28 日期间,发作于 1 例以上患者的不良事情。一些患者有多起不良事情。由于并无适用于高敏肌钙蛋白(心脏生物标志物)血清水平缓血脂水平的不良事情分级规范,因而表中列出了数值与基线比较发作恶化的患者份额。惯例医治组中高敏肌钙蛋白升高的患者份额高于洛匹那韦-利托那韦组(14.1% vs. 9.5%)。惯例医治组 55 例患者(52.4%)和洛匹那韦-利托那韦组 65 例患者(68.4%)的血脂水平在归入实验时正常,但归入实验后反常。一切逝世均由呼吸衰竭引起。ARDS 标明急性呼吸困顿综合征。
评论
此项随机实验发现,关于 COVID-19 重症患者,与独自选用惯例支撑医治比较,在惯例医治的基础上加用洛匹那韦-利托那韦并未呈现临床情况改进或逝世率下降。但是,在扫除了 3 例前期逝世患者的改进意向医治剖析中,两组间至临床情况改进的中位时刻(中位数,15 天 vs. 16 天)尽管差异很小但到达了统计学显着性。有必要留意一下的是,本实验的总逝世率(22.1%)显着高于 COVID-19 住院患者初始描述性研讨中陈述的 11%~14.5% 的逝世率,这标明咱们归入的是重症患者人群。
本实验的患者人群在入组时的患病时刻和疾病严峻程度方面具有异质性;过后剖析发现,在症状呈现后 12 日内承受医治的亚组中,临床情况敏捷恢复(16.0 日 vs. 17.0 日)且逝世率较低(19.0% vs. 27.1%),而症状呈现后 12 日之后承受医治的亚组则没有这一情况(图 S2A和 S2B)。对 COVID-19 前期采纳洛匹那韦-利托那韦医治是否具有临床好处是一个需求进一步研讨的重要问题。这一调查成果与有研讨标明 SARS-CoV-2 病毒性肺炎在发病第 2 周发作发展共同,也与之前在 SARS 和严峻流感的抗病毒研讨中调查到的起效时刻共同。
此外,咱们发现在承受洛匹那韦-利托那韦医治的患者中,发作严峻并发症(急性肾损害和继发感染)或因呼吸衰竭而需求无创或有创机械通气的人数少于未承受医治的患者。咱们可依据这些调查成果提出假定,且需求经过进一步研讨来确认在疾病某一阶段用药的洛匹那韦-利托那韦可否削减 COVID-19 的某些并发症。
与独自选用惯例支撑医治比较,咱们并未发现加用洛匹那韦-利托那韦可下降病毒 RNA 载量或缩短检出病毒 RNA 的时刻。实验结束时(第 28 天),洛匹那韦-利托那韦组有 40.7% 的患者仍可检出 SARS-CoV-2 RNA。近期一份陈述指出,COVID-19 重症患者排出病毒的中位持续时刻是 20 天,最长可达 37 天 24。上述研讨和本研讨中均无依据标明洛匹那韦-利托那韦具有显着抗病毒作用。药物看似未显现抗病毒作用的原因没有清晰,但本实验的采样办法很或许欠佳。
本实验仅仅间断性采样(在第 1、5、10、14、21 和 28 日采样),在前 5 日进步采样频率有或许更具体地体现出两组在这一关键时期的病毒载量动力学特征。此外,之前的研讨标明,口咽拭子样本的病毒载量低于鼻咽拭子样本,而且重要的是,咱们无法对下呼吸道分泌物进行采样。有必要留意一下的是,依据所用的细胞类型不同,洛匹那韦关于 SARS-CoV 的体外对折有用浓度(EC50)规模为4.0~10.7 μg/mL,但也有其他研讨陈述洛匹那韦无效或需求更高浓度(25 μg/mL)才干发作按捺作用。关于 MERS-CoV,EC50 值规模为 5~7 μg/mL。成人服用洛匹那韦后的均匀峰值(9.6 μg/mL)和谷值(5.5 μg/mL)血清浓度都仅仅挨近上述浓度。EC50 值是否是适宜的阈值,以及人体血浆内的非结合态洛匹那韦浓度是否足以按捺 SARS-CoV-2 仍有疑问。
承受洛匹那韦-利托那韦医治的患者中有近 14% 无法完结 14天 的完好阶段。底子原因是厌食、厌恶、腹部不适或腹泻等胃肠道不良事情和两起严峻不良事情(均为急性胃炎)。有 2 例承受洛匹那韦-利托那韦的患者发作了自限性皮疹。肝损害、胰腺炎、更严峻的皮疹和 QT 延伸危险等副作用以及因 CYP3A 按捺而发作多种药物相互作用的或许性在上述联合用药中均有充沛记载。本实验中调查到的副作用令人担忧以改进患者结局为意图而进步洛匹那韦-利托那韦剂量或延伸用药时刻的做法。
本实验有几项局限性。其间特别重要的是本实验未选用盲法,因而知晓分组情况或许影响了临床决议计划,从而或许影响了咱们选用的 7 分等级量表。咱们将持续随访这些患者,评价其远期预后。两组患者的基线特征根本平衡,但洛匹那韦-利托那韦组的咽喉部病毒载量略高,因而该组的病毒仿制或许较多。
尽管咱们在两组间并未调查到并用的其他药物(例如糖皮质激素)的用药率有差异,但这或许是别的一个稠浊要素。此外,在洛匹那韦-利托那韦组中,第 14 日和第 28 日时别离有约 45% 和 40% 患者的口咽拭子 RNA 检测成果呈阳性,但咱们并不知晓是否仍有具有感染性的病毒,由于咱们并未测验别离病毒,也未评价是否或许会呈现了对洛匹那韦易理性下降的 SARS-CoV-2 变异株。最终,咱们并无这些重症患者(其间许多为危重患者)的洛匹那韦露出水平数据。
总归,咱们发现在 COVID-19 重症患者中,洛匹那韦-利托那韦未能显着加快临床情况的改进,未能下降逝世率,也未能削减咽喉部检出的病毒 RNA。这些前期数据将为未来展开研讨评价该计划和其他药物对 SARS-CoV-2 感染的作用供给辅导。SARS 医治中曾将洛匹那韦-利托那韦与其他抗病毒药联用,现在也正在 MERS-CoV 中研讨联合用药,而在 COVID-19 医治中将洛匹那韦-利托那韦与其他抗病毒药联用可否增强抗病毒作用,改进患者临床结局仍有待确认。
附件图表
表S1. 改进意向医治人群的结局。
表中依据情况对人数(百分比)或中位数(四分位距)进行了总结。
缩写:ICU = 重症监护病房;HFNC = 经鼻高流量氧疗;IMV = 有创机械通气;ECMO = 体外膜氧合;TTCI = 至临床情况改进的时刻。
临床情况改进(事情)的界说为在修订版 7 分等级量表上,临床情况改进 2 分或许患者出院。
* 这一总数不包含在随机分组后 24 小时内逝世,因而未承受洛匹那韦/利托那韦医治的 3 例患者。
§ 差异标明为率差或中位数差(Hodges-Lehmann估量)和置信区间。
† 使用 Cox 份额危险模型预算危险比。
表S2. 改进意向医治人群的病毒学结局。
经过定量逆转录聚合酶链反应(qRT-PCR)剖析法确认的 mITT 剖析集(不包含未检出病毒 RNA 的患者)的 AUC。小于 PCR 检测法定量下限而且大于定性检测限的成果用1 log10 仿制/mL 添补;病毒 RNA 阴性的患者成果用0 log10 仿制/mL添补。AUC,曲线下面积。
§ 差异标明为率差或许 Hodges-Lehmann 估量值差异和 95% 置信区间。
图S1. 改进意向医治人群中至临床情况改进时刻的Kaplan Meier曲线。
图S2. 意向医治人群中依照患病时刻(≤ 12 天[图 A] vs. > 12 天[图 B])制作的至临床情况改进时刻的 Kaplan Meier 曲线。
图S3. 意向医治人群中依照疾病严峻程度(NEWS2 评分 > 5 分[图 A] vs. ≤ 5 分[图 B])制作的至临床情况改进时刻的 Kaplan Meier 曲线。
图S4. 意向医治人群中至临床情况恶化时刻的 Kaplan Meier 曲线。
图S5.意向医治人群中第 1、7、14 和 28 日时首要结尾分类的份额散布。
图S6. 改进意向医治人群中依照患病时刻(≤ 12 天[图 A]vs. > 12 天[图 B])制作的随时刻推移,经过 qPCR 检测的咽拭子(病毒阳性人群)SARS-CoV-2 病毒 RNA 载量相关于基线的改变。
本文首发于大众号:NEJM医学前沿(ID:NEJM-YiXueQianYan),授权丁香园发布
责任编辑:
